
Summary
Tumor initiation is characterized by a loss of normal cell identity and function. Moreover, during disease progression cancer cells undergo further phenotypic transformations in response to changes resulting from microenvironment and therapy-induced selective pressures. The given adaptability of cancer cells contributes to multiple aspects of tumor progression, including tumor initiation and metastasis, and resistance to chemotherapy. Changes in cellular phenotype can be triggered by genetic alterations, but very often, they are the result of transcriptional and epigenetic reprogramming. By elucidating the molecular mechanisms by which cancer cells exploit plasticity, we aim to identify new vulnerabilities in cancer and improve patient survival.
Major Research Goals
- Characterize different layers of transcriptional regulation defining cancer cell identity and specific cancer phenotypes
- Explore molecular mechanisms underlying therapeutic resistance in basal PDAC and NSCLC
- Identify novel epigenetic vulnerabilities in basal PDAC and NSCLC
- Exploit biologically relevant cancer models for studying cellular and phenotypic plasticity in basal PDAC and NSCLC
Highlights
Transcriptional programs determining cellular identity are dominated by the activity of transcription factors (TFs) that recognize and bind specific cis-regulatory DNA regions in the genome, i.e. enhancers and thereby regulate target gene expression. Identity-specific enhancers harbor specific chromatin landmarks and are transcriptionally active. Non-coding enhancer RNAs (eRNA), in turn, contribute to transcriptional regulation of target genes by mediating promoter-enhancer chromatin looping or serve as scaffolds for transcriptional activators and chromatin remodelers (Fig.1). In addition, the cellular identity at a given time point is largely influenced by signals from the tissue environment. Most of the information delivered by signaling pathways converge on enhancers via activation and recruitment of signaling-induced TFs that cooperate with lineage-specific TFs to maintain identity-specific transcriptional programs.
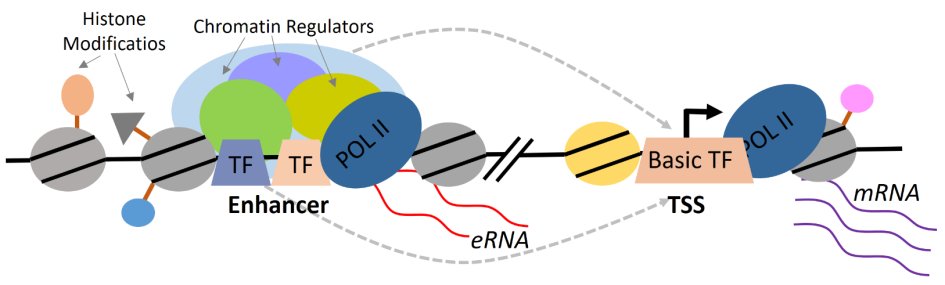
Figure 1. Transcriptional layers of identity-specific gene expression regulation.
The master TFs bind cooperatively to enhancer DNA elements and recruit coactivators and the transcription apparatus to mediate active chromatin state. These enhancer elements produce enhancer RNAs (eRNAs) that can in addition mediate enhancer-promoter looping to stimulate transcription of target genes.
Our major scientific focus over past couple of years evolved around the investigation of molecular and epigenetic mechanisms controlling lineage-specific transcription and gene regulatory patterns involved in cell fate determination in differentiating osteoblasts (Karpiuk et al., 2012; Najafova et al., 2017; Baumgart et al., 2017; Najafova et al. 2021). Our previous work identified that epigenetic activation of OB-specific lineage markers relied on RNF20/RNF40-driven histone H2B monoubiquitination (Karpiuk et al., 2012). Histone H2B monoubiquitination is an active histone mark that facilitates transcriptional elongation of genes. Furthermore, by utilizing conditional knockout mouse models we were able to demonstrate that RNF40-driven H2B monoubiquitination has a stage-dependent function in controlling osteoblast differentiation and is essential for bone cell crosstalk via regulation of a vitamin D-mediated transcriptional program (Najafova et al., 2021).
In a parallel effort, we examined and characterized the role of bromodomain protein 4 (BRD4) – dependent epigenetic reprograming in differentiating OBs (Najafova et al., 2017). BRD4 is an epigenetic reader protein and performs the role of transcriptional activator and modulates gene expression by regulating RNA Polymerase II elongation. The genome-wide localization analyses of BRD4 followed by transcriptome-wide analysis of gene expression changes revealed that despite the described general function in transcriptional regulation, BRD4 exerts a selective role in regulating lineage-specific transcriptional programs. The mechanistic investigation of this phenomenon suggested that BRD4-driven context-dependency is largely defined by enhancer programs that require BRD4 to maintain the lineage identity.
Epigenetic reprogramming and loss of cellular identity are also critical steps during pancreatic ductal adenocarcinoma (PDAC) development. PDAC is one of the most aggressive solid malignancies characterized by high phenotypic plasticity. Two major subtypes of PDAC have been identified that display clear differences in clinical behavior. While classical PDAC exhibits a less aggressive phenotype, basal PDAC poorly responds to the standard chemotherapy and displays dismal prognosis in patients. Basal PDAC is characterized by a squamous-like gene expression program and tends to show more metastatic behavior. The aberrant expression of squamous lineage markers in PDAC has been invariably associated with expression and activity of ΔNp63, a master regulator of the normal squamous lineage. By utilizing comprehensive multi-omics approach, we verified that ΔNp63 specifies cell identity in basal PDAC and that it does so via dynamic regulation of the epigenetic landscape (Fig.2). Moreover, we identified a basal subtype-specific transcribed enhancer program (B-STEP) which provides tumor cells with high epithelial-to-mesenchymal (EMT) plasticity and metastatic phenotype (unpublished data). Moreover, by application of chromatin topology analysis we identified a ΔNp63-dependent super enhancer in basal-like A PDAC which regulates the expression of the SNAI2 transcription factor, one of the important mediators and inducers of EMT (unpublished data) (Fig.3). Notably, we were able to detect B-STEP eRNAs in primary patient histological tumor samples, thereby demonstrating a proof of principle that subtype-specific eRNA detection could potentially serve as a diagnostic tool for patient therapy stratification and monitoring.
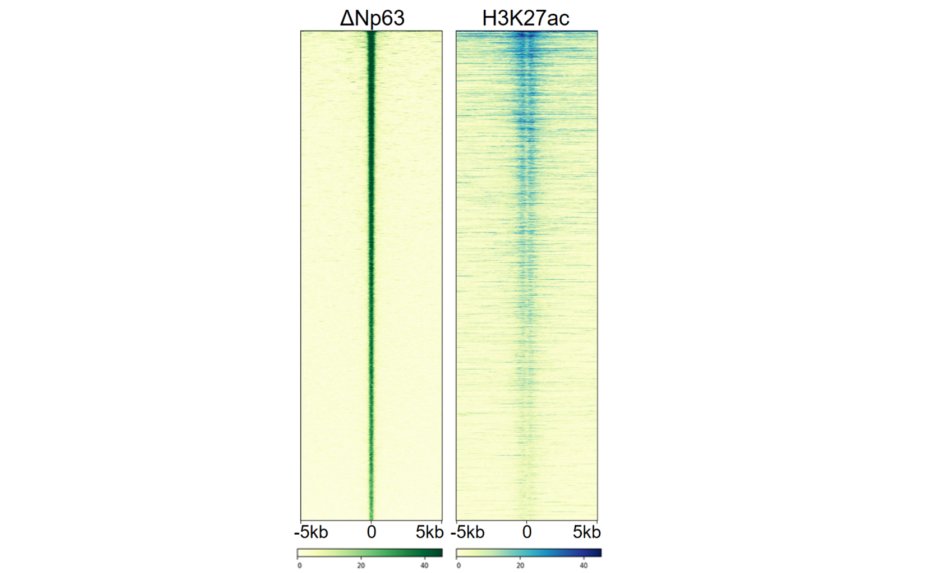
Figure 2.
By utilizing PDX model from a patient with basal PDAC we performed ChIP-Seq and identified that TP63 bound regions harbour active chromatin mark H3K27ac linked with promoter and enhancer regions of active genes.
Outlook
Over the next years, the major focus of our research at the Robert Bosch Center for Tumor Diseases will be centered on the exploration and understanding of transcriptional regulatory programs determining cell identity and plasticity in solid epithelial carcinomas. Our main efforts will be dedicated to the analysis of the different layers of transcriptional regulation controlling specific cancer cell phenotypes with the aim to subsequently exploit these programs for therapeutic interventions (Fig.4). To test and validate our hypotheses we will exploit different – omics approaches on biologically relevant cell culture systems (primary and immortalized), patient-derived primary tumor material and translational models (PDX) obtained via partners at the clinic.
SNAI2 as determinant of aggressive phenotype in PDAC
While initially focusing on ΔNp63-driven basal PDAC, we will build upon our previous findings to elucidate the role of SNAI2 transcription factor in supporting the aggressive PDAC phenotypes. SNAI2 is a known regulator of EMT and usually its overexpression is indicative of poor prognosis for PDAC patients. To study how and whether the transcription factor activity of SNAI2 is required for driving oncogenic gene expression programs in PDAC, we will initially utilize various cell culture models of PDAC with aggressive behavior. Namely, by using CRISPR-mediated locus-specific knock-in approach to deliver dTAG sequence into SNAI2 gene locus we will examine how rapid loss of SNAI2 protein affects the PDAC phenotype by performing different phenotypic characterization assays. Moreover, to examine the underlying molecular mechanisms dictated by SNAI2, we will determine gene expression (RNA-sequencing), epigenetic and enhancer activity (ChIP-, ChRO-seq, HiChIP) changes induced by rapid loss and gain of SNAI2 protein in the same setting. Furthermore, we will perform SNAI2 ChIP-seq to identifiy the binding sites of SNAI2 in PDAC and will in parallel correlate these to the changes induced in epigenetic landscape by SNAI2. Via the bioinformatics approaches, we will predict and validate the potential interacting partners of SNAI2 and their role in maintenance of oncogenic transcription in PDAC and test their potential for therapeutic targetability. Our previous work identified SNAI2 as a direct target of ΔNp63 in basal-like A PDAC, however SNAI2 can be independently induced via different signaling cascades in other subtypes of PDAC. We therefore will aim to determine the molecular differences between SNAI2 function in ΔNp63-proficient and ΔNp63-deficient PDAC subtypes by performing comprehensive bioinformatic analysis. Finally, to investigate SNAI2-dependent tumor cell intrinsic mechanisms and interactions with tumor microenviroment we will utilize single cell and/or single nuclei RNA-seq in primary tumor and patient derived xenograft samples.
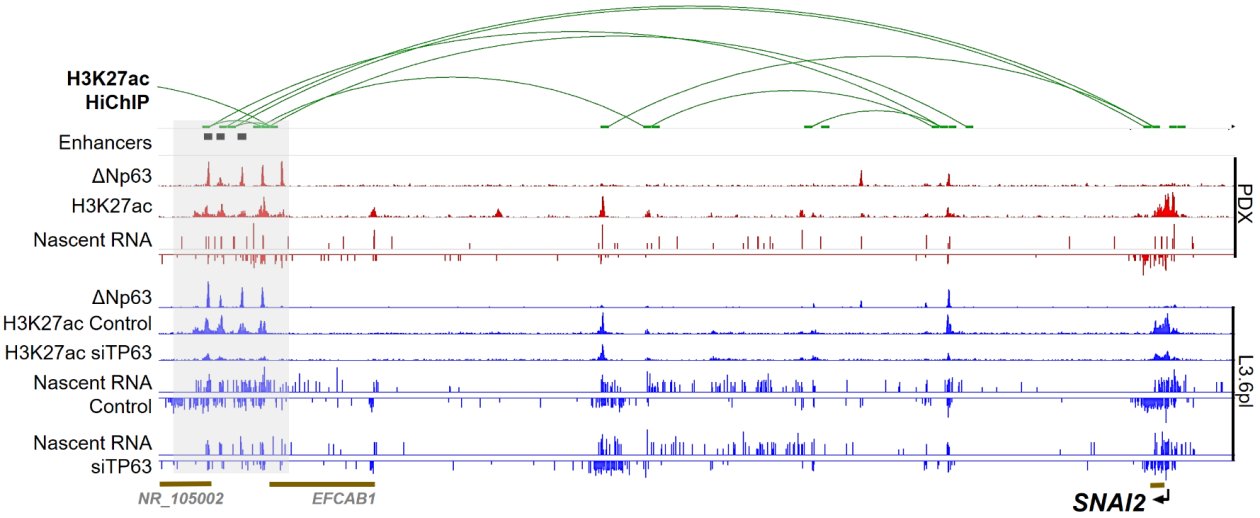
Figure 3. Binding profile of ΔNp63 at downstream enhancers of SNAI2 in a basal PDAC. H3K27ac HiChIP and nascent RNA capture sequencing were used to identfy these enhancers in basal PDX (red) and cell line (L3.6pl, blue) models.
The essentiality of identified enhancer regions for transcriptional regulation of SNAI2 gene was validated via epigenetic silencing approach utilizing dCas9-KRAB system.
Analysis of ΔNp63-driven oncogenic program in NSCLC
Given that aberrant overexpression of the ΔNp63-dependent program is a defining feature of squamous cell carcinomas of different origins, we will aim to delineate the ΔNp63-dependent molecular regulatory pathways responsible for squamous cancer development and progression using as a model non-small cell lung carcinoma (NSCLC). Namely, we will use NSCLC cell culture models to examine the function of ΔNp63 and its association with other transcription factors in regulating the epigenetic landscape and enhancer activity driving the lung squamous cell carcinoma identity. We will utilize dTAG degron system (Nabet B et al., 2018) to achieve rapid and selective degradation of P63 protein in order to study the direct role of ΔNp63 in driving the squamous gene expression program and subsequently on the phenotypic behavior of the cell lines. We will complement this with high-throughput ChIP-seq technique to evaluate the changes in epigenetic landscape as well as mRNA- and nascent RNA-seq to examine the gene expression and enhancer activity alterations accompanying the acute loss of P63 function. Subsequently, based on investigation of ΔNp63-driven changes we will aim to predict and subsequently validate the P63 interacting TFs that support P63 in mediating oncogenic program in lung squamous cell carcinoma. In addition, to identify novel epigenetic regulators involved in ΔNp63-driven gene expression maintenance we will use loss of function CRISPR-Cas9 epigenetic screening libraries established in NSCLC cell models and test the effect of pharmacological inhibition of these on cancer progression and chemotherapy response. This in turn will further our understanding of the biology of ΔNp63- driven squamous lung carcinoma and provide advances in diagnosis, prognosis and therapy.
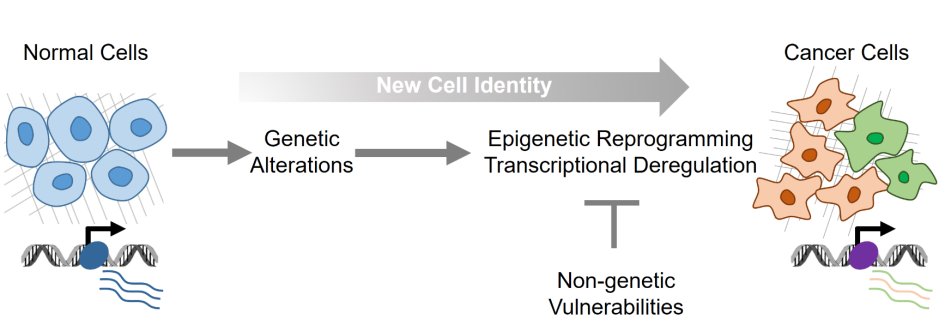
Figure 4. Cancer cells rely on deregulated epigenetic and transcriptional programs to maintain their identity.
Understanding the key components on which the dysregulated transcriptional programs depend in cancer cells is essential for identifying new therapeutic targets. Such transcriptional dependencies can be identified through focused mechanistic studies of gene control programs functioning in both normal and neoplastic cells.