
Summary
Pancreatic ductal adenocarcinoma (PDAC) is one of the most aggressive malignancies with a very dismal prognosis. While genetic changes play a central role in pancreatic cancer development, these changes ultimately elicit their long-term effects via changes in cell identity due to altered gene expression. Notably, pancreatic cancer displays a very high degree of plasticity whereby cellular identity can rapidly change in a non-genetic manner in response to environmental signals or selective pressure. Our group investigates the molecular epigenetic mechanisms controlling gene transcription and therapy response in pancreatic cancer.
Major Research Goals
- Identify and characterize novel regulators of pancreatic cancer cell plasticity
- Exploit targetable transcriptional and epigenetic mechanisms controlling pancreatic cancer subtype identity and therapy response for patient treatment
- Develop and utilize relevant patient-centered experimental models for pancreatic cancer
- Test novel mechanism-based therapeutic approaches as a basis for future clinical studies
Highlights
While molecular subtyping has led to dramatic improvements in patient treatment for many other types of cancer, only recently have different molecular subtypes been identified in pancreatic cancer. In the case of PDAC, different subtypes have been hypothesized to originate from distinct cells of origin, as is likely the case for other tumors with similar histological subtypes such as esophageal adenocarcinoma and squamous cell carcinoma. However, recent data exploring the genomes of different tumor clones from the same patient with distinct subtype morphologies and gene expression revealed a common origin of classical and basal PDAC. Based on these findings, it can be concluded that a high degree of plasticity between PDAC phenotypes is primarily driven by epigenetic mechanisms (since genomic changes are minimal or absent). Importantly, these findings also suggest a subtype hierarchy whereby the basal subtype emerges from the classical subtype and the basal program supersedes the classical (Figure 1). Thus, we hypothesize that a better understanding of the interplay between the subtype-specific programs will uncover mechanisms to reverse the basal program and restore chemotherapeutic sensitivity in highly aggressive PDAC.
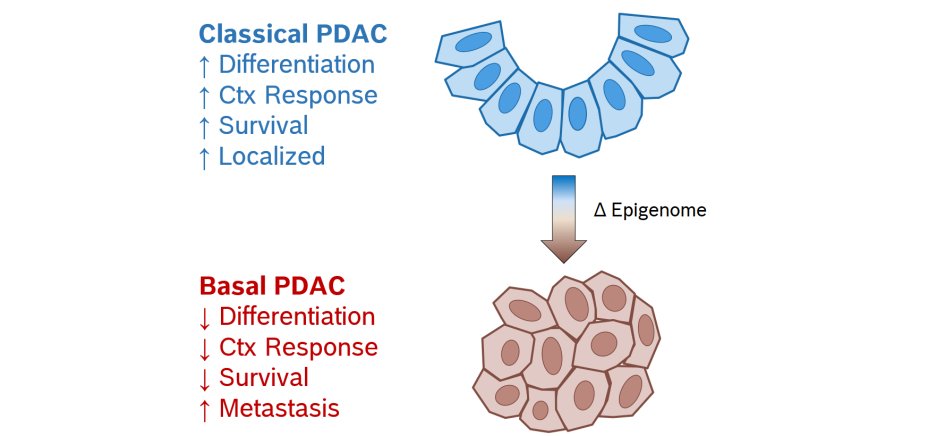
Figure 1. Pancreatic cancer subtypes.
Pancreatic ductal adenocarcinoma (PDAC) can be separated into two major molecular subtypes termed classical and basal, which display distinct outcomes, morphologies and therapeutic responsiveness. Studies suggest that both have a common origin and can even co-exist within a given patient.
Our ongoing work not only confirms our previous findings (Hamdan et al., PNAS 2018) that multiple transcription factors (p63, BHLHE40, HIF1A and RXRA) colocalize to subtype-specific enhancers in basal-like PDAC cells (Figure 2). Importantly, we have utilized genome-wide chromatin interaction studies, coupled with epigenome mapping, transcription factor binding and transcriptome-wide gene expression studies, to identify putative subtype-specific enhancer regions controlling the aggressive basal-like PDAC subtype. The functional importance of these could be confirmed by epigenome editing using a nuclease-deficient Cas9 coupled to a KRAB repressor domain (Figure 3).
In other ongoing studies the Johnsen group is examining the mechanisms of therapeutic resistance in pancreatic cancer. This work significantly expands on the group’s recent work examining resistance to gemcitabine (Kutschat et al., Canc Res 2021) and identifies novel enhancer networks that can be effectively targeted to restore sensitivity to standard of care chemotherapy (Hamdan et al. Gut 2023). It is envisaged that this work will serve as a basis for future early phase clinical trials combining these novel treatment combinations (see press release).
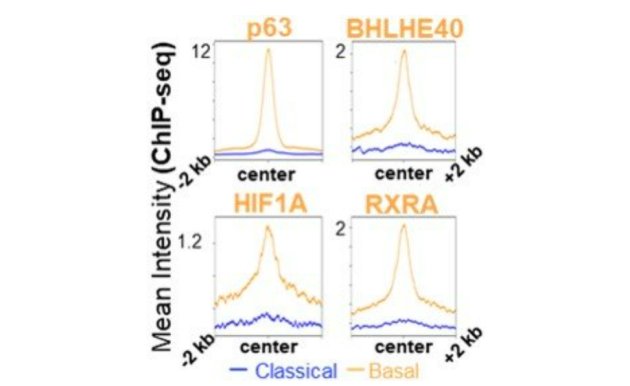
Figure 2. Colocalization of transcription factors in basal PDAC.
Aggregate plots showing occupancy of B-MTFs at basal vs classical specific peaks in L3.6pl
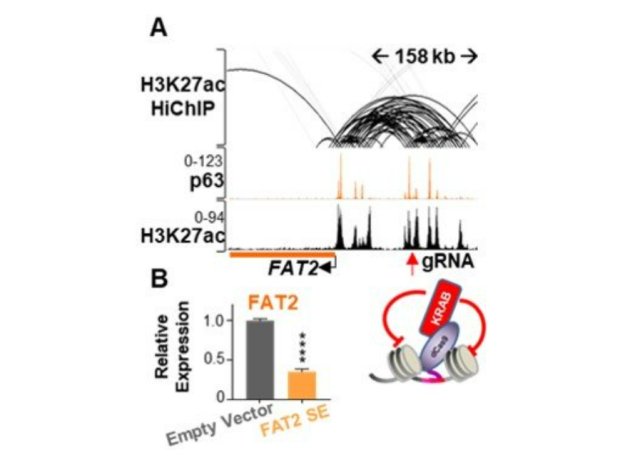
Figure 3. Enhancer essentiality.
(A) Binding profiles of p63 and H3K27ac at FAT2 gene and SE with Hi-ChIP showing high frequency of interaction between the two elements. The red arrow shows the site of recruitment of dCas9-KRAB leading to SE silencing. (B) Gene expression of FAT2 mRNA upon silencing of FAT2 SE.
Outlook
At the Robert Bosch Center for Tumor Diseases, the Johnsen group will continue to intensively investigate various transcriptional and epigenetic mechanisms controlling pancreatic cancer cell phenotype and therapy responsiveness. The group will also utilize functional genomics approaches (loss of function screens) to identify and characterize novel regulators of PDAC cell identity and test these for their potential therapeutic potential. Importantly, together with various clinical departments at the Robert Bosch Hospital (Pathology, Surgery, Gastroenterology, Oncology), we will continue to investigate and test our hypotheses in primary clinical samples with the goal of providing a foundation for future clinical studies.
Building upon our previous findings (Hamdan & Johnsen, 2019; Mishra 2017a; Mishra 2017b; Patil 2020), we will continue to investigate the transcription factors driving specific molecular subsets of PDAC, identify and characterize transcriptional and epigenetic cofactors controlling their activity and determine how these can be utilized for clinical translation by studying the effects of their perturbation in clinically relevant translational model systems.
Specifically, using targeted degradation (AID, dTAG), knockdown and CRISPR/Cas9-based genome editing approaches, combined with our expertise in next generation sequencing-based approaches, we will determine the importance of specific factors in controlling PDAC cell fate and determine their potential utility for targeting in therapeutic intervention strategies.
For example, our group has been interested in the role of Bromodomain and Extraterminal (BET) proteins such as BRD4 and BRDT for several years (Liu 2021; Wang 2021; Nagarajan 2017; Najafova 2017). There are numerous clinical candidate small molecule BET inhibitors currently in clinical trials, as well as a number of new generation, bromodomain-specific BET inhibitors currently in development. Our work will continue to investigate the utility of these inhibitors in PDAC with relation to molecular subtype and therapy response and examine the genomic mechanisms by which they elicit such effects.
Similarly, we will utilize our extensive epigenetic datasets to interrogate other potential targetable molecular pathways. For instance, in addition to epigenetic regulators like the BET proteins, we are currently investigating a select set of signaling pathways as well as enzymes and protein complexes involved in different stages of gene transcription and RNA processing as potential targets for PDAC treatment.
Using these multiple modern molecular biology approaches we aim to not only characterize general molecular mechanisms controlling gene regulation, but uncover viable options for the clinical translation of these findings for the treatment of pancreatic cancer.