
Summary
Blood homeostasis is sustained by the continuous and dynamic production of hematopoietic cells within specific lineages. Disruptions in the regulatory mechanisms of this process can result in malignant hematopoiesis, ultimately leading to the onset of hematological malignancies. Technology advances have enabled the identification of recurrent gene variations and dysregulation in hematological malignancies. Many of these alterations are linked to transcriptional regulation programs that drive the initiation, progression, or relapse of the diseases. Our research focuses on unravelling the intricate gene regulatory networks that underlie the pathogenesis of human leukemia. We employ state-of-the-art techniques, including multiom-ics profiling, high-throughput screens, gene gain- or loss-of-function, and genome or epigenome editing, conducted on leukemia cell lines, primary patient samples, and mouse tumor models. Our primary objective is to identify novel molecular biomarkers and therapeutic targets or approaches for the treatment of human leukemia.
Major Research Goals
- Examine the role of RNA molecule and RNA binding proteins in transcriptional regulation through chromatin remodelling in leukemia
- Investigate epigenetic mechanisms determining the dysregulation of key oncogenes in leukemogenesis
- Characterize and target leukemia-associated oncoproteins for tumor eradication
Highlights
Acute Myeloid Leukemia (AML) is a hematopoietic malignancy characterized by pathologic expansion of myeloid cells without terminal differentiation. It is the most common acute leukemia in adult and is highly aggressive, with ~70% of patients succumbing to the disease. AML is considered a paradigm of transcriptional dysregulation in cancer, as recurrent mutations associated with AML were found to commonly hit transcription factors (TFs), epigenetic modifiers and genome structure proteins (Figure 1). These mutations could alter the transcriptome through remodelling epigenetic environment and three-dimensional (3D) chromatin structure, converging on the dysregulation of gene networks to trigger malignant hematopoiesis.
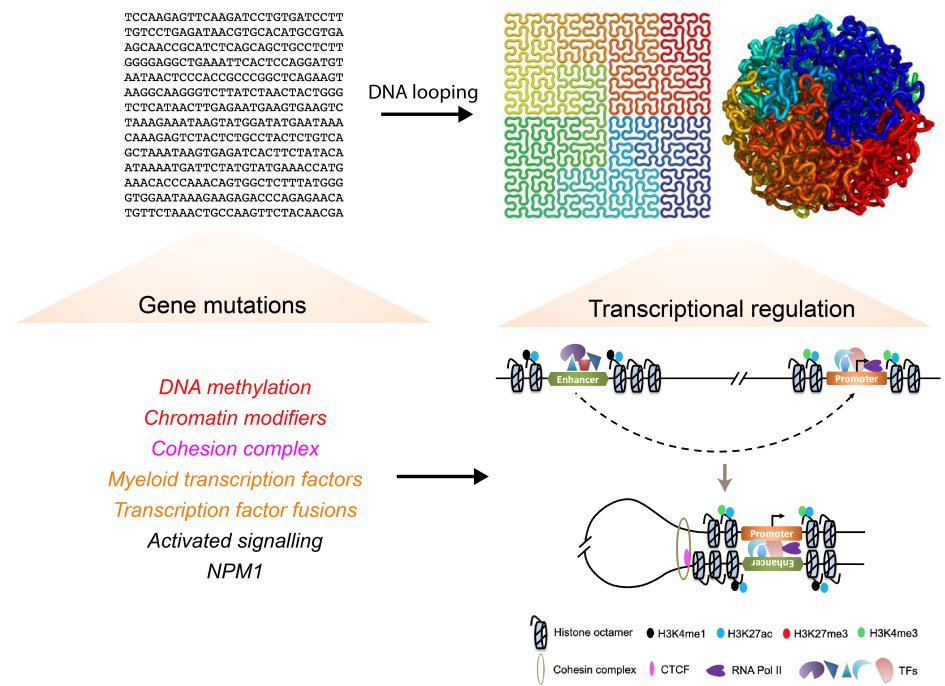
Figure 1. Gene mutations and dysregulations involving both the linear and spatial genome of AML.
In AML, DNA mutations frequently hit genes that are categorized into groups with diverse function. Transcriptional regulation involves chromatin modifications and 3D remodelling between enhancers and promoters.
Our initial research effort involved the identification of genetic aberrations with prognostic impact in myeloid malignancies, as well as the investigation into molecular mechanisms underlying the induction or maintenance of myeloid malignancies caused by gene alterations. Our work identified the dysregulated TFs including MN1 (Heuser et al. 2011) and IRF8 (Sharma et al. 2015), as well as the epigenetic regulator MLL5 (Yun et al. 2014) in the transcriptional control of AML-related gene network. We further contributed to the characterization of engineered mouse or cell models with deficiency in chromatin remodellers such as CBP (Horton et al. 2017), UTX/UTY (Gozdecka et al. 2018), EZH2 (Basheer et al. 2019) and cohesin complex members (Sasca et al. 2019), which are all commonly mutated in hematopoietic malignancies. Interestingly, loss of any of these epigenetic players could rewrite transcription programs through modulating enhancer functions, highlighting the crucial role of enhancer dysregulations in leukemogenesis.
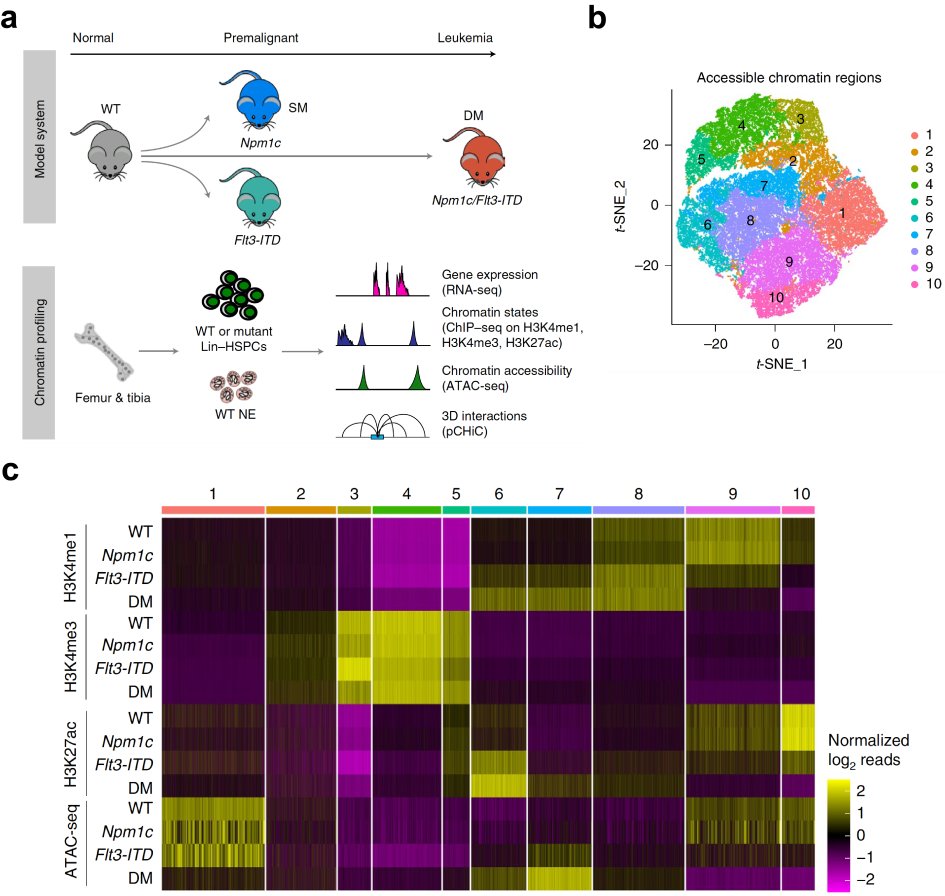
Figure 2. Gene mutations synergize 3D enhancer remodelling upon leukemia induction.
a, schematic of study design. b and c, t-SNE plot (b) and heatmap (c) showing clustering of cis-regulatory elements based on chromatin profiles across four cellular states.
Meanwhile, we applied an integrated multi-omics approach to study the epigenetic changes at 3D genome levels in the hematopoietic stem and progenitor (HSPC) compartment in a leukemia evolution model, utilizing a murine allelic series carrying the two most common mutations in AML, the Flt3-ITD and Npm1c (Yun et al. 2021; Figure 2a). We identified clusters of cis-regulatory elements exhibiting dynamic enhancer or promoter activity induced by mutations (Figure 2b,c). This analysis facilitated the capture of novel distal regulatory events responsible for the dysregulation of leukemia critical genes. An exemplar was the identification of a novel superenhancer sitting ~1Mb upstream of Hoxa genes cluster, regulating the transcription of Hoxa genes through chromatin interaction in Npm1c-mutated HSPCs (Figure. 3a). CRISPR-Cas9-mediated genetic disruption of this superenhancer resulted in downregulation of Hoxa genes and impairment of leukemia cell propagation (Figure 3b-d). Taken together, these data demonstrated that enhancer dysregulation symbolized epigenetic changes at higher-order chromatin level in leukemogenesis, and manipulation of altered enhancers represented a novel approach to target leukemia.
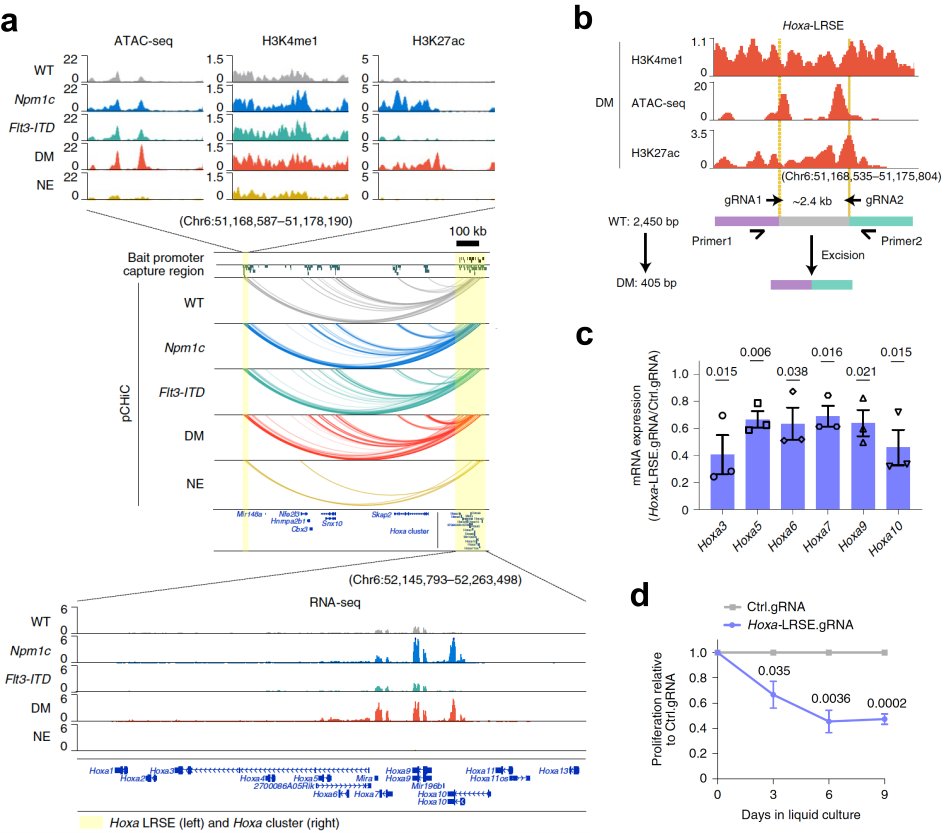
Figure 3. Identification of Hoxa long range superenhancer (Hoxa-LRSE) in Npm1 mutant HSPCs. a, integrated chromatin profiles and 3D interactions at LRSE and Hoxa cluster in HSPCs and neutrophils (NE). b, Hoxa-LRSE disruption using CRISPR-Cas9 plus dual gRNAs. c, relative mRNA expression of Hoxa genes upon Hoxa-LRSE deletion. d, proliferation assays of double mutant leukemia cells upon Hoxa-LRSE disruption in comparison to control.
Outlook
At Robert Bosch Center for Tumor Diseases, we will continue our dedication to the translational cancer research in the field of hematology and aim to expand our knowledge in understanding the multi-layered gene regulatory networks in hematological malignancies.
Previously we identified extensive bidirectional transcriptional signatures within a subset of leukemia-activated enhancers. The production of enhancer RNAs (eRNAs) is intricately linked to altered enhancer accessibility and modification, enhancer-promoter looping and targe gene dysregulation. These observations suggest that enhancer transcription might be a novel layer of epigenetic regulation through 3D enhancer remodelling. We therefore hypothesised that dysregulated eRNAs are essential to maintain leukemia propagation, through modulating chromatin at 3D level, and might be targetable for the eradication of leukemia cells. We will employ multiple genome-wide approaches to identify dysregulated eRNAs that are critical for leukemia maintenance, and investigate the underlying molecular mechanisms (Figure 4). Eventually we would evaluate the functional importance and clinical relevance of our candidate eRNAs in human AML samples, aiming to translating our finding into clinical practice.
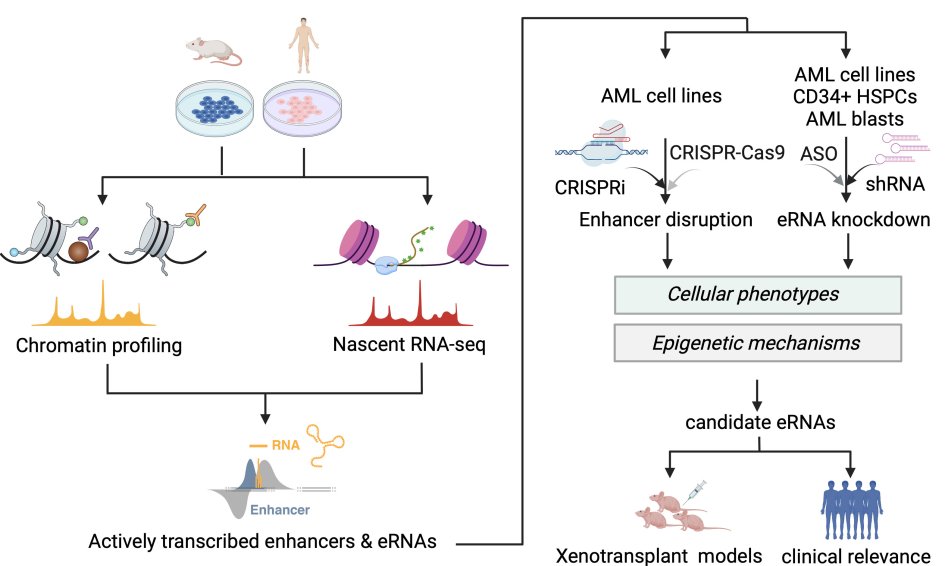
Figure 4. Experimental procedures for the identification and characterisation of dysregulated eRNAs in leukemia.
Besides, we will endeavour to explore the epigenetic mechanisms controlling the dysregulation of key oncogenes associated with malignant hematopoiesis. These oncogenes include FLT3 and HOXA9, which are frequently mutated or overexpressed in AML, respectively. We will apply high-throughput profiling and screening methods on AML cell lines, primary samples and leukemia cell differentiation models, with expectation to capture bona fide TFs and cis-regulatory elements driving oncogene overexpression in leukemia cells. The results will assist the discovery of new targets for treating these specific types of leukemia.
Our other initiative will focus on characterizing and targeting leukemia-associated
oncoproteins for tumor eradication. In this part, we would dissect the role of mutant NPM1 (NPM1c) in chromatin remodelling. Using engineered fluorescence labelled NPM1c cell model, we will investigate how NPM1c triggers nuclear chromatin redistribution, and identify epigenetic regulators participating in this process. We will examine the possibilities of targeting these regulatory factors in NPM1c leukemia mouse models and patient samples. In addition, we will perform loss-of-function screens to find out gene networks responsible for the depletion of NPM1c in AML cells.