
Summary
Hallmarks of cancer include sustained proliferative signaling and resistance to cell death. Proliferation is induced by receptors in the plasma membrane that activate kinases and transcription factors to induce expression of pro-proliferative genes. Thus, mutations affecting activity or expression of signaling proteins are frequent in tumors. However, no single mutation is sufficient for malignant transformation because fail-safe mechanisms exist, e.g. apoptosis - a cellular suicide program. Consequently, defective apoptosis signaling opens the route to malignant transformation. The group investigates (dys)regulation of apoptosis and signaling pathways in cancer cells to identify new therapeutic concepts. Elucidating molecular mechanisms instigates investigator initiated trials. Principally, we aim to translate basic research into clinical application to improve anti-cancer therapy.
Major Research Goals
- Elucidate molecular mechanisms underlying cell death induction and identify new drug targets and synergies by generating and utilizing molecularly defined tumor models
- Provide new concepts for anti-cancer therapy by using advanced fluorescence imaging, knock-out screens, next-generation sequencing and single cell analyses
- Broaden the applicability of therapeutic modalities by applying elucidated mechanisms and molecular understanding of signaling pathways
- Translate molecular mechanisms into clinical application to improve therapeutic options, based on biological or genetic markers
Highlights
BH3-mimetics targeting anti-apoptotic BCL-2 family proteins.
The mitochondrial apoptosis pathway is regulated by the BCL-2 family of proteins. The BCL-2 family is comprised of pro- and anti-apoptotic subgroups harboring at least one of the four different BCL-2 homology domains (Fig. 1). The anti-apoptotic “BCL-2 like” proteins interact via a hydrophobic groove with the BH3-domain of pro-apoptotic siblings. When BH3-only proteins block the hydrophobic groove of BCL-2 like proteins, restriction of the effector proteins BAX, BAK, and BOK is released and allows their oligomerization in the outer mitochondrial membrane. Effector oligmoerization mediates release of cytochrome c inducing formation of the apoptosome and activation of apoptosis specific proteases, the caspases. Caspases cleave substrate proteins and dismantle the cell. However, in a recent study, the group described direct involvement of mitochondria rather than BCL-2 proteins in resistance to cisPlatinum induced cell death (Kleih et al. 2019).
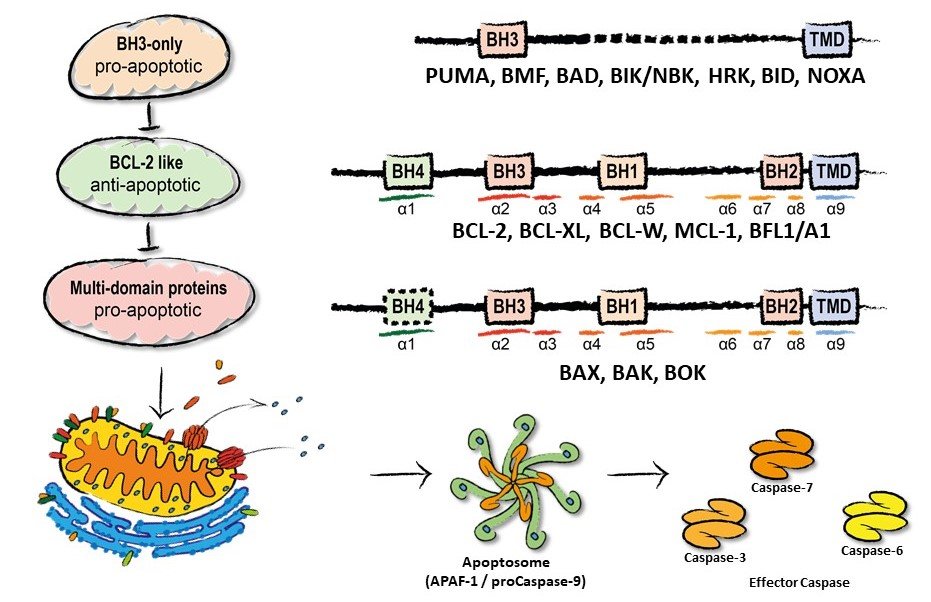
Figure 1. The BCL-2 family of proteins and intracellular signalling cascade.
Therapeutic BH3-mimetic drugs ABT 199/Venetoclax (top), S63845 (middle), A1155463 (bottom) that block anti-apoptotic BCL-2, MCL-1, and BCL-xL, respectively
Current projects build on insights into the mechanisms of BCL-2 family interaction and development of small molecule BH3-mimetics that specifically inhibit anti-apoptotic BCL-2 proteins. The BCL-2 inhibitor ABT-199/Venetoclax is effective in multiple myeloma, acute myelogenous leukemia and chronic lymphatic leukemia. Resistance to ABT-199 is frequently mediated by overexpression of MCL-1 and can be overcome by the MCL-1 inhibitor S63845. Sarcomas, a rare group of malignant tumors in soft tissue, are highly heterogeneous complicating the search for therapeutic options. In a drug screen in different sarcoma cell lines we identified efficacy of combination of ABT-199 (Venetoclax) and Bortezomib (BTZ, Velcade), clinical approved drugs. BTZ blocks protein degradation by the proteasome. ABT-199 and BTZ synergistically induce apoptosis in sarcoma cells from patients and cell lines. Apoptosis induction depends on BAX and the MCL-1 antagonizing BH3-only protein NOXA (Muenchow et al. 2020). Intriguingly, the BH3-mimetic ABT- 199 transcriptionally activates expression of PMAIP1/NOXA (Weller et al. 2022). Thus, ABT-199 has a double impact on apoptosis induction by simultaneously blocking i) BCL-2 and releasing BAX and ii) MCL-1 releasing BAK. Proteasome inhibition reduces NOXA degradation and exacerbates apoptosis induction (Fig. 2). The BCL-2 independent pro-apoptotic activity of ABT-199 represents a generally active principle of a tumor-agnostic mechanism that might offer alternative therapeutic strategies for anti-cancer treatment. Importantly, this is supported by experiments in hepatocellular carcinoma cell lines and slice cultures: NOXA expression is relevant for the efficacy of cell death induction by ABT-199/Venetoclax combined with the death-receptor agonist TRAIL (Busche et al. 2021).
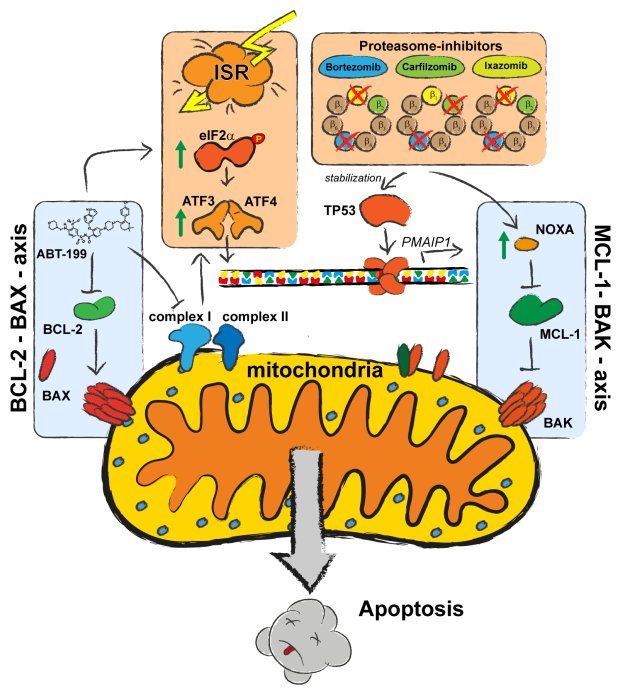
Figure 2: Schematic of the elucidated regulatory mechanisms contributing to synergistic apoptosis induction by ABT-199 and Bortezomib.
New concepts of BCL-2 family regulation
Cells are sentenced to death when the anti-apoptotic capacity of “BCL-2 like” proteins is exhausted and oligomerization of pro-apoptotic multidomain effector proteins BAX, BAK, and BOK (Einsele- Scholz et al. 2016) mediates release of cytochrome c from mitochondria (Fig. 1). As shown for BAX, oligomerization involves substantial conformational changes allowing the interaction of α-helices α2/α3 in the mitochondrial membrane and α4/α5. Effector dimers then form higher order oligomers that until now are structurally not characterized in detail. Interestingly, BOK is localized to the endoplasmic reticulum where it is involved in calcium transfer to mitochondria. Localization of BCL-2 proteins to subcellular membranes is mediated by C-terminal transmembrane domains (Fig. 3). The TMDs are also proposed to be involved in oligomerization of effectors BAX and BAK. Thus, the BH3- and transmembrane domains impact on localization and interaction of BCL-2 proteins (Stehle et al. 2018). We investigate the interaction and function of individual BCL-2 protein TMDs by e.g. advanced fluorescence microscopy techniques (Fig. 3) with the aim to develop new strategies to modulate the BCL-2 network. Elucidation of hitherto unresolved molecular mechanisms might indicate new therapeutic approaches for drug mediated cell death induction or inhibition by intercepting TMD interaction.
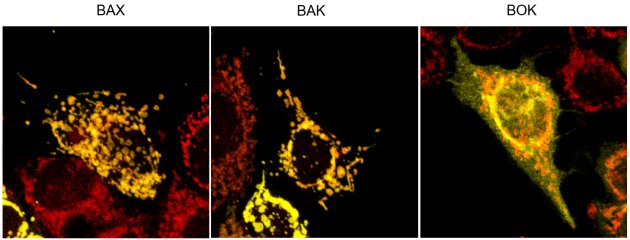
Figure 3. Subcellular localization of the fluorescently labelled TMDs (yellow) of effector proteins BAX, BAK, and BOK relative to mitochondria (red).
NSCLC and Inhibitors targeting receptor tyrosine kinases.
Extracellular signals that regulate proliferation are transduced by receptors in the plasma-membrane that activate signaling pathways to induce target gene expression. Signal transduction frequently involves phosphorylation (Grondona et al. 2020) and culminates in the translocation of transcription factors from cytosol into the nucleus (Keminer et al. 2019). An important class of receptors are receptor tyrosine kinases, which include various growth factor (GF) receptors: epidermal (EGF), nerve (NGF), platelet derived (PDGF), vascular endothelial (VEGF), fibroblast (FGF), and insulin- like (IGF). Mutation of RTKs is prominent in non-small cell lung cancer (NSCLC). Approximately 15% of Caucasian and up to 50% of Asian advanced NSCLC harbor driver mutations of the epidermal growth factor receptor (EGFR). The understanding of EGFR as a receptor tyrosine kinase (RTK) in molecular detail has enabled the development of tyrosine kinase inhibitors (TKIs) that specifically block cancer-driving EGFR variants and TKI-based therapy is efficient in EGFR-mutant NSCLC. Similarly, targeted therapy exists for tumor entities with driver mutations in other RTKs, e.g. ALK (5%), MET (3%), ROS1 (2%), HER2 (2%), RET (2%) (Fig. 4). On the downside of targeted therapy is the apparently inevitable evolution of resistance and tumor recurrence. To overcome therapy- induced resistance caused by EGFR-mutation, TKIs are successively adapted. In case of EGFR, 3rd generation TKI Osimertinib is active against common therapy-induced on-target mutations. First line therapy with Osimertinib shows prolonged (deeper) response and enhanced progression free survival, however, at the cost of extremely limited options for follow-up therapies. Thus, strategies that are effective in therapy-induced resistant tumors or regimens that prevent tumor escape are highly demanded. Tumors escape from TKI-therapy by: i) secondary mutation of RTKs, ii) mutations in downstream signaling molecules, or iii) mutation in parallel signaling pathways. Together with the Department of Molecular Oncology of the Robert Bosch Hospital (Prof. H.-G. Kopp) we investigate the combination of existing TKIs with approved inhibitors for pro-survival proteins to enhance sensitivity of NSCLC cells to apoptosis induction and thereby induce a deeper response to diminish tumor cell escape.
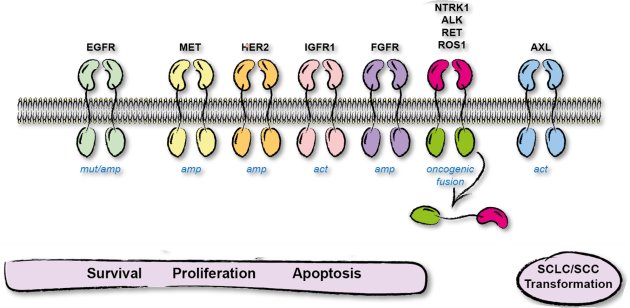
Figure 4: Receptor tyrosine kinases signal via kinase cascades to regulate cell sruvival, proliferation, and apoptosis.
Dysregulation of receptor tyrosine kinases by overexpression or mutation is identified as driver of tumorigenesis. Drugs, that specifically inactivate mutant RTKs for targeted therapy are in clinical application.
In case of tumor cells with mutation of the RTK ALK sensitivity to the ALK inhibitor Alectinib is enhanced by photodynamic therapy (Gillissen et al. 2021) whereas resistance to TKI is overcome by combined application of TKI and proteasome inhibitors (PI). TKI+PI induce expression of the pro-apoptotic BCL-2 protein NOXA that is causally involved in cell death induction. We found that NOXA induction is crucial for synergistic cell death induction by PIs in combination with the BCL-2 inhibitor ABT-199 (Muenchow et al. 2020). Thus, we investigate co-treatment of RTK-mutant cells with BH3-mimetic drugs as a rational approach to overcome resistance to or enhance therapeutic efficacy of (R)TKIs. Apart from RTKs, in NSCLC the tumor suppressor PTEN is frequently mutated or deleted thereby limiting the employment of personalized therapy, as it drives resistance to established targeted therapies like EGFR antagonists. However, tumors harboring a loss of function mutation in PTEN can be therapeutically addressed by irradiation in combination with ATM inhibition (Fischer et al. 2022).
Outlook
We elucidated the molecular mechanism mediating synergistic cell death induction of ABT-199 & proteasome inhibitors (Muenchow et al. 2020; Weller et al. 2022). We will broaden the analyses to additional cancer entities and samples from cancer patients to extend and stratify the applicability of the identified therapeutic approach. We will analyze the role and tissue/tumor-specific expression of BH3-only proteins (e.g. BIM, NOXA, PUMA) and anti-apoptotic BCL-2 proteins by protein analysis and RNA-seq to establish (surrogate) markers. We plan to pinpoint the relevance of BCL-2 family and unrelated proteins for sensitivity to combinatorial therapy approaches by utilizing i) BH3-mimetic drugs and ii) CRISPR/Cas9-mediated knock-out screens. The goal is to build a basis for the tissue- unspecific identification of markers for sensitivity to BH3-mimetic drugs and combinations with e.g. kinase inhibitors, proteasome inhibitors (Weller et al. 2022), DNA-damaging drugs (Kleih et al. 2019) or chromatin-modulating drugs (von Tresckow et al. 2019) in order to identify new modalities of anti- cancer treatment.
To deepen the response of NSCLC to targeted therapy with RTKIs we investigate the combined application of BCL-2 inhibitors to enhance sensitivity to apoptosis induction. To achieve a further understanding of the molecular premises we will investigate expression of BCL-2 family proteins and their relevance and regulation during TKI induced cell death. These analyses will be performed in established and newly generated cellular model systems of NSCLC with specific resistance mediating mutations of EGFR or activated parallel survival pathways. Again, we will apply clinically approved drugs (TKIs, BH3-mimetics) to establish markers for the stratification of molecular determinants of therapy response. To identify alternative drug targets we will perform knock-out screens in characterized model systems. The findings will be challenged in CDX models and markers will be verified/falsified in PDX models and patient derived tumor tissue. The results will enable investigator initiated trials and perspectively offer therapy modalities beyond osimertinib and classic chemotherapeutic drugs by preventing development of resistance to targeted therapy.
New insights in transmembrane domain mediated subcellular localization and interaction of BCL-2 proteins instigate approaches to modulate the BCL-2 network via a so far unrecognized route. These basic research activities are envisioned to offer new routes to cell death induction and completely novel therapeutic approaches for anti-cancer treatment by e.g. drugs that block membrane targeting of BCL-2 proteins or disrupt TMD interaction. Also, coupling of existing BH3-mimetics to moieties that interact specifically with TMDs might enhance specificity or therapeutic window of the bi-specific drugs.